
Hola a todos y todas.
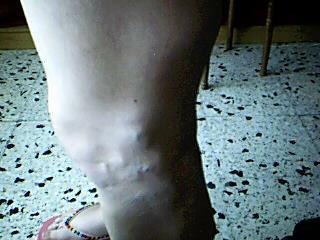
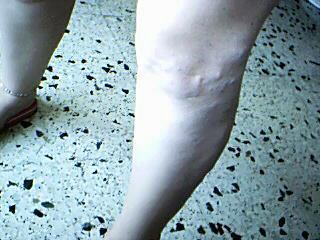
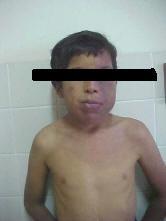
Con pesar veo que mi búsqueda no produce frutos, y también me encuentro ante una situación contradictoria ya que, han aparecido personas que tienen el Síndrome o bien lo padece algún familiar.
Desde aquí os propongo que, por favor, hagáis uso del grupo que creé para ello.
Este blog es informativo, hablo de la enfermedad y hablo sobre lo que a mí me pasa. Pero siempre que encuentro a alguien, recibo el mensaje en mi correo electrónico. No es que me moleste, pero tengo varias direcciones y no siempre estoy en ellas. Tengo la principal, pero ahí no llegan los mensajes.
Por favor, hacer uso de este grupo que creé para hablar y comunicarme con vosotros/as.
Dándole al enlace que dejo aquí abajo, cada nuevo miembro que se una, lo sabré de inmediato ya que llegará la información a mi correo principal, y además, contribuiréis a formar un grupo estable, donde podamos conocernos todos, ya que hasta la fecha soy yo la que os conozco uno a uno, y para mejorar las experiencias de todos, intercambiar nuestros casos, dudas, inquietudes, no hay nada mejor que hacerlo desde este grupo.
Aquí os dejo el enlace, y por favor, hacer uso de él. Gracias. Silvia.
Con pesar veo que mi búsqueda no produce frutos, y también me encuentro ante una situación contradictoria ya que, han aparecido personas que tienen el Síndrome o bien lo padece algún familiar.
Desde aquí os propongo que, por favor, hagáis uso del grupo que creé para ello.
Este blog es informativo, hablo de la enfermedad y hablo sobre lo que a mí me pasa. Pero siempre que encuentro a alguien, recibo el mensaje en mi correo electrónico. No es que me moleste, pero tengo varias direcciones y no siempre estoy en ellas. Tengo la principal, pero ahí no llegan los mensajes.
Por favor, hacer uso de este grupo que creé para hablar y comunicarme con vosotros/as.
Dándole al enlace que dejo aquí abajo, cada nuevo miembro que se una, lo sabré de inmediato ya que llegará la información a mi correo principal, y además, contribuiréis a formar un grupo estable, donde podamos conocernos todos, ya que hasta la fecha soy yo la que os conozco uno a uno, y para mejorar las experiencias de todos, intercambiar nuestros casos, dudas, inquietudes, no hay nada mejor que hacerlo desde este grupo.
Aquí os dejo el enlace, y por favor, hacer uso de él. Gracias. Silvia.
 <-- Haced clik aquí
<-- Haced clik aquí













{kind=link}
{kind=link}